
Electrochemical Deposition of Hydroxyapatite (HAP) Coating
All samples of hydroxyapatite were prepared via the electrochemical depositional process described in the literature of Professor Yates’ group at the University of Rochester, with the exception of two samples which were plasma sprayed hydroxyapatite coatings on titanium substrates; these were purchased from a common supplier for comparison to electrochemically deposited HAP.
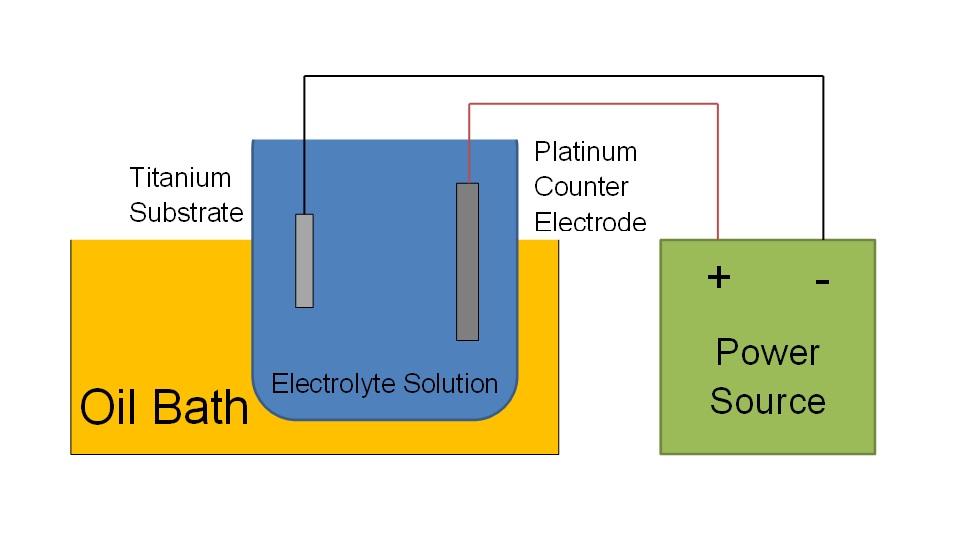
The hydroxyapatite layer was electrochemically deposited on titanium plates (12.5 by 12.5 by 0.89mm) which were washed thoroughly with hand soap, rinsed, then washed with an industrial detergent solution, and thoroughly rinsed again. The substrate was cleaned ultrasonically for 2 hours in 50/50 ethanol/acetone and then for 30 min in DI water. The cleaned titanium plate was used as the anode for electrochemical deposition of hydroxyapatite and the cathode was a platinum plate (25 by 25 by 0.127 mm). The anode and cathode were maintained parallel to one another at a fixed distance of separation of 10 mm using a custom Teflon holder. The electrodes were connected to a power supply using platinum wire for the cathode and silver wire for the anode. The electrodes were then submerged in an aqueous solution consisting of 1.25 mM CaCl2 and 0.827 mM K2HPO4. The solution’s electrolytic ability was enhanced with the addition of 138 mM NaCl; the solution’s pH was buffered with tris(hydroxymethyl)-aminomethane and adjusted to pH 7.2 with concentrated HCl acid. The electrolyte solution was maintained at ~95ºC using an oil bath. Both the electrolyte solution and oil-bath were stirred using magnetic stir bars. Deposition was carried out for 5 minutes at 12.5 mA/cm2 (area relative to the platinum plate); the coated substrate was then removed from the solution, allowed to cool, then rinsed thoroughly with DI water. The sample was allowed to dry in air. Two HAP samples were adhered to a sample stub by a small piece of conductive carbon tape for imaging.
 |